Основы медицинской генетики
Медицинская генетика является составной, но самостоятельной частью генетики человека и так же, как общая генетика, изучает закономерности наследственности и причины изменчивости, но в отношении не нормальных, а патологических признаков организма. Важной задачей медицинской генетики является изучение причин возникновения наследственной патологии человека, характера ее наследования в семьях, распространенности в различных популяциях. Медицинская генетика исследует природу наследственной патологии на функциональном, клеточном и молекулярном уровнях, разрабатывает методы профилактики, ранней диагностики и лечения наследственных болезней.
В последнем десятилетии наблюдается увеличение относительной доли генетически обусловленных заболеваний в патологии человека. Это частично связано с улучшением диагностики, но в значительной мере — с ухудшением экологической обстановки, усилением мутагенеза и накоплением патологических мутаций. По приблизительным подсчетам, в мире рождается в год более 2 миллионов детей с тяжелыми формами наследственных болезней. Смертность детей с наследственными заболеваниями до пятилетнего возраста в 4-20 раз выше, чем общая смертность детей.
Проблема здоровья человека теснейшим образом связана с наследственностью. Измененная программа развития, измененная норма реакции на обычные факторы внешней среды являются причинами наследственной патологии.
3.1. Наследственная и врожденная патология
Наследственная патология — это патология, обусловленная нарушениями (патологическими мутациями) наследственного аппарата (хромосом, генов) половых клеток родителей и передающаяся через них следующим поколениям.
Патологическая мутация может быть свежей — происходить в половых клетках родителей и являться причиной рождения детейГлава 3. Основы медицинской генетики
с наследственной патологией у родителей, в семьях которых данная патология ранее не встречалась. Возникнув, патологическая мутация может передаваться из поколения в поколение в семье. Время возникновения патологических мутаций можно установить, проследив проявление и передачу аномального признака в родословной (генеалогический метод генетического исследования).
Наследственная патология как результат реализации в определенных условиях среды измененной генетической программьтожет проявиться в любые сроки онтогенеза (чаще всего — в раннем детстве, но и в зрелом возрасте, например, хорея Гентингтона, и даже пожилом, например, подагра).
Презентация на тему: Медицинская реабилитация в психиатрии
... регламентации временных параметров реабилитационных мероприятий не всегда возможна. Проведение медицинской реабилитации больных с психическими расстройствами базируется на следующих основных ... жизнь. Первоначальная типология включает три вида реабилитации: медицинскую, профессиональную и психосоциальную. *Медицинская реабилитациявключает в себя фармакотерапию, лечебную физкультуру, диетическое ...
Врожденная патология — это всякое пренатально обусловленное отклонение от нормального строения органа или системы органов. Грубо выраженные аномалии, приводящие к частичной или полной функциональной непригодности органа или создающие у человека косметическую проблему, принято называть врожденными пороками развития. Незначительные отклонения, совместимые с нормальным функционированием и не обезображивающие внешний облик, укладываются в понятие мелких дисплазий или стигм дизэмбриогенеза.
Клинические проявления врожденных пороков разнообразны. По распространенности пороки делят на изолированные, системные и множественные. Изолированные пороки локализуются в одном органе (пороки сердца, почек и др.), системные пороки объединяют несколько пороков в одной системе органов (пороки развития нервной системы, мышечной, лимфатической и др.), множественные пороки составляют группы аномалий в двух и более системах органов. В большинстве случаев у детей с врожденной патологией регистрируются изолированные пороки (80%), реже наблюдаются множественные и системные.
Доля врожденной патологии в структуре заболеваемости составляет 6-10%, смертности — 15% (среди новорожденных этот показатель достигает 40%).
У нас в стране ежегодно рождается более четверти миллиона детей с тяжелыми врожденными пороками развития.
Врожденные пороки могут быть наследственными, являться следствием мутаций, а могут быть результатом эмбриотоксического действия целого ряда факторов внешней среды.
Не являются наследственными врожденные аномалии, если они обусловлены повреждающим действием вредных факторов
JI. А. Попова, Т. П. Степанова. Основы генетики в коррекционной педагогике
(тератогенов) на генетически нормальный зародыш. В этих случаях наблюдаются нарушения развития органов, полностью копирующие эффект мутагена (фенокопирование).
Характер пре- натального нарушения зависит от сроков беременности и критических периодов развития организма: так, пороки сердца и сосудов формируются в критические для этой системы 4-5-ю недели беременности, нервной системы и лицевого скелета — на 4-5-й и 6- й неделе соответственно.
Мутагенно и тератогенно обусловленная врожденная патология клинически и анатомически проявляется сходным образом. Большую часть (85-90%) составляют широко распространенные изолированные пороки: врожденный вывих бедра (1% от общего числа новорожденных), косолапость (1-0,6%), расщелины губы и неба (2,7%).
Реже встречаются множественные пороки, в основном хромосомного происхождения (например, множественные пороки скелета, конечностей, внутренних органов и т. п.) и системные (например, альбинизм и глухота, пороки опорно-двигательного аппарата, нервной системы и т. п.).
По Международной классификации болезней, наследственные болезни и врожденные пороки развития отнесены к одной общей рубрике, частота врожденных пороков сильно варьируется в отдельных странах от 0,15 до 15% от числа всех новорожденных. По данным ВОЗ, средняя частота рождения детей с аномалиями составляет 3%.
Развитие моторики у детей с интеллектуальной недостаточностью
... и биологических факторов. Актуальность темы обусловлена тем, что проблема психомоторного развития детей дошкольного возраста с интеллектуальной недостаточностью стала привлекать внимание ученых относительно ... действие или движение человека регулируется определенными звеньями сложной функциональной двигательной системы. В основе ее современной концепции лежат положения учения И.П. Павлова ...
Установление конкретной причины и механизма повреждения в каждом отдельном случае является сложной задачей (если учесть многообразие средовых факторов, их взаимодействие, явления фено- и генокопирования и др.).
Возникновение любого патологического признака есть результат сложного взаимодействия наследственного предрасположения и внешнесредовых влияний. Но в практических целях, в зависимости от ведущего этиологического фактора, выделяют:
- мутагенные пороки (наследственные), связанные с мутациями на геномном, хромосомном или генном уровнях;
- тератогенные пороки (экзогенные), обусловленные действием повреждающих факторов на эмбрион (эмбриональный период — от 16-го дня после оплодотворения до конца 10-й недели) или плод (плодный период — от 11-й недели до родов);
Глава 3. Основы медицинской генетики
3) мультифакториальные пороки, вызванные сочетанием генетических и тератогенных эффектов, при этом изолированно ни один из них не является причиной порока.
Мутагенным и тератогенным действием обладают различные факторы внешней среды: физические (ионизирующая радиация, ультрафиолетовое излучение, токи и звуки сверхвысокой частоты, механические воздействия и др.); химические (сельскохозяйственные, промышленные, бытовые химические вещества, алкоголь, никотин и наркотические средства, некоторые лекарственные препараты, гормоны); биологические (вирусы, микробы, простейшие и др.).
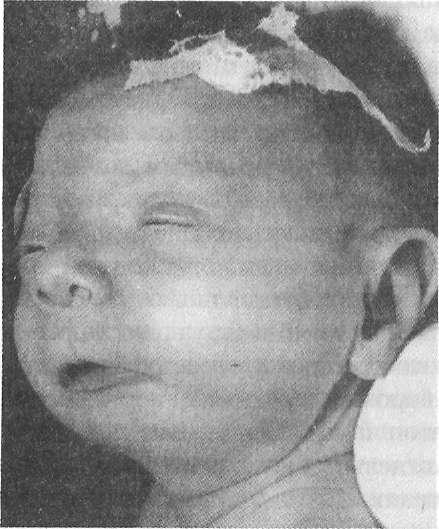
Рис. 3.1. Особенности лица при феталь- ном алкогольном синдроме (Цит. по: Козлова С. И. и др. Наследственные синдромы и медико-генетическое консультирование. — М.: Медицина, 1987.)
Хорошо известен мутагенный и тератогенный эффект всех видов ионизирующей радиации (альфа-, бета-, гамма-, ультрафиолетового и рентгеновского излучения), внешнее проявление которого зависит от длительности и вида излучения, индивидуальной чувствительности и состояния организма на стадии внутриутробного развития, на котором происходит облучение. Сходным действием об
ладают другие физические факторы, такие, как высокие и низкие температуры, различные вибрации, механические воздействия и др.
Тератогенное и мутагенное действие обнаружено у ряда химических веществ, содержащихся в предметах бытового назначения (клей для мебели, стенные панели и полы, выделяющие токсические вещества при эксплуатации).
Особенно опасно применение таких средств и «зараженного» оборудования в детских садах, школах и других местах коллективного пребывания детей. Среди химических веществ особую группу составляют алкоголь и наркотики в связи с высокой
JI. А. Попова, Т. П. Степанова. Основы генетики в коррекционной педагогике
вероятностью тератогенных эффектов с тяжелой клинической картиной. У новорожденных она описывается как алкогольный феталь- ный синдром с характерным сочетанием симптомов: микроцефалия, эпикант, поперечные складки шеи, гипоплазия нижней челюсти, длинный фильтр, часто системные пороки внутренних органов, скелета и др. (рис. 3.1).
Основные факторы развития личности
... являются хорошей основой для оправдания этноцентрического, националистического сознания, однако не могут оправдать решающего влияния физического фактора на развитие личности. Действительно, ... в сходных физических и географических условиях формируются различные типы личностей, и, наоборот, очень часто бывает так, что схожие групповые признаки ...
Многие лекарственные вещества и медикаменты, рекомендуемые беременным, могут быть небезвредными для развивающегося организма. Например, прием некоторых антибиотиков на 6-й неделе беременности приводит к окрашиванию и гипоплазии эмали не только молочных, но и постоянных зубов ребенка и даже к тяжелым порокам развития. К числу таких веществ относятся гормональные препараты, транквилизаторы, цитостатики и др. Обычно такие медикаменты назначаются ограниченно, но в ряде случаев речь идет о широком применении. Так, небезызвестный тяжелыми последствиями талидомид за годы использования (около 6 лет) был продан в Европе в количестве 1,6 тонны.
Из биологически обусловленных тератогенных факторов следует особо выделить инфекционные агенты периода внутриутробного развития: вирусы краснухи, кори, гриппа, эпидемического паротита, цитомегалии, а также простейшие токсоплазмы. Вирус краснухи способен сам индуцировать первичные пороки развития органов и тканей с характерным сочетанием нарушения зрения, слуха, гидро- и микроцефалии, развивающимися более чем в 20% случаев заболевания матери в первом триместре беременности.
Особенности этиопатогенетических механизмов наследственных и внешнесредовых врожденных аномалий обусловливают трудности дифференциальной диагностики между ними и актуальность решения этой задачи. Эта проблема непосредственно касается семей, где появились дети с врожденными пороками. Существенное значение имеет оценка мутагенного и тератогенного вклада внешнесредовых факторов в механизм и клинику отдельных нозологических форм в масштабных профилактических целях. Поэтому при использовании всех известных методов генетики особое значение имеет тщательное изучение фенотипических признаков в большом числе родословных и установление закономерностей, общих для всей совокупности.
Одной из таких закономерностей является семейное накопление случаев болезни. Повторные случаи заболевания в семье — характерный
признак болезней с наследственной предрасположенностью. По распределению этих болезней среди родственников пробанда устанавливают тип их наследования. Однако при малом размере семей наследственная патология может проявиться только у одного члена семьи. С другой стороны, семейный характер болезни еще не доказывает ее наследственного характера, т. к. это может быть связано с социально-средовыми и другими внешними факторами.
Важным критерием дифференциальной диагностики между этими типами врожденной патологии является количество признаков диспластичности, называемых также дизморфогенетическими или дизонтогенетическими признаками, или малыми пороками развития. Наличие одного-пяти диснластических признаков (стигм) может быть результатом экзогенных воздействий или генных мутаций, а сочетание их, превышающее в сумме семь-восемь, свидетельствует о высокой вероятности хромосомных нарушений.
Общее количество диспластических признаков насчитывает несколько сотен, они разделяются на стигмы черепа, лица, ушей, шеи, туловища, конечностей, кисти и стоп, глаз (см. приложение).
Факторы роста и развития в постнатальном онтогенезе
... гормоны наряду с другими факторами, вызывают многие важные сдвиги в морфофункциональной организации подростка. Они регулируют развитие первичных и вторичных половых признаков, влияют на половое ... намечаются половые различия в развитии жироотложения (выше у девочек) и начинается изменение формы тела. Перепубертатный (лат. «pubertalis» – возмужалость) этап в развитии человека является доминирующим, ...
Например, диспластическими признаками ушей является их низкое расположение, асимметричность, оттопыренность, большие отвисающие уши, маленькие деформированные уши и т. д. К стигмам глаз относятся: эпи- кант, узкая глазная щель, маленькое глазное яблоко, монголоидный и антимонголоидный разрез глаз, глубоко посаженные глаза и т. д.
Многочисленными клиническими наблюдениями установлено, что некоторые гены, контролирующие отдельные признаки лица, ушных раковин и т. д., оказывают определенное влияние и на центральную нервную систему (плейотропное действие гена).
Мутантное состояние этих генов приводит к фенотипическим аномалиям: косой разрез глаз, эпикант, гипертелоризм, низкое расположение ушных раковин, низкая линия роста волос, плоский затылок — и достоверно коррелирует с нарушениями функций центральной нервной системы.
Нужно иметь в виду, что стигмы дизэмбриогенеза, являясь показателями неблагоприятного течения пренатального периода развития, указывают на возможность нарушений во внутренних органах. Эти негрубые структурные аномалии значительно облегчают возникновение функциональных расстройств в том или ином органе или системе. Причем этот морфофункциональный «фон» сопровождает человека на протяжении всей жизни.JI. А. Попова, Т. П. Степанова. Основы генетики в коррекционной педагогике
Отмечается также связь множественных фенотипических аномалий с пониженной устойчивостью к инфекциям, особенно вирусным. Это обусловлено тем, что несколько тысяч генов, контролирующих работу иммунной системы, располагаются не только в шестой хромосоме, но и рассеяны по многим другим. Наличие большого числа аномальных фенотипических признаков указывает на диффузное поражение генома и рассредоточенных генов иммунной системы с последующим ослаблением иммунитета. Возникающие при этом хронические процессы развиваются как следствие постоянного действия мутантного гена или хромосомного дисбаланса, либо как результат взаимодействия генетических и средовых факторов при болезнях с наследственной предрасположенностью. Для последних характерно и рецидивирующее течение, связанное как с особенностями генотипа, так и с воздействием средовых факторов.
Закономерно, что для фенотипически аномальных индивидуумов характерна пониженная жизнестойкость. К возрасту 40-45 лет, а иногда и раньше, у них, казалось, прежде здоровых, вдруг возникает какая-либо патология, которая принимает хроническую форму. На ее фоне проявляются другие, якобы самостоятельные болезни, и к 55-60 годам накапливается целый «букет», состоящий из 12-15 болезней. Кроме того, именно у фенотипически аномальных лиц чаще развиваются различные осложнения при инфекционных заболеваниях, хирургических вмешательствах, использовании лекарственных препаратов.
У лиц с фенотипическими аномалиями отмечается пониженная репродуктивная способность, что, по-видимому, поддается аналитической трактовке с позиций диффузного нарушения генома.
Таким образом, традиционное деление этиологических факторов на экзо- и эндогенные, на средовые и наследственные имеет несколько условный характер. В действительности любое средо- вое воздействие осуществляется на фоне неповторимого генотипа данного индивида, и любой генотип реализуется в определенных средовых условиях. Большинство врожденных аномалий человека детерминированы многочисленными и разнообразными как средовыми, так и наследственными факторами, т. е. являются мультифакториальными (тератогенные факторы провоцируют переход наследственной предрасположенности в порок развития).
Типологические особенности в эмоциональном развитии детей с умственной ...
... младшего школьного возраста. 3. Осуществить сравнительное изучение эмоционального развития детей с умственной отсталостью и задержкой психического развития в различных возрастных группах. 4. Выделить возрастные ... эмоциональные результаты своей деятельности. Именно в дошкольном возрасте ребенок осваивает высшие формы экспрессии - выражение чувств с помощью интонации, мимики, пантомимы, что ...
Эти пороки встречаются с частотой 1:1000. К аномалиям такого
Глава 3■ Основы медицинской генетики
рода относятся: врожденный вывих бедра, косолапость и конская стопа, орофациальные расщелины, анэнцефалия, врожденные пороки сердца и др.
Отдельные элементы этих аномалий (дисплазия бедра, орофациальные микропризнаки и др.) могут возникать и прослеживаться в родословной, передаваясь как доминантные, рецессивные или сцепленные с полом признаки. Накопление таких единичных микропризнаков обозначается как мутационный груз. Наличие многочисленных аномалий (стигм, дисплазий) у носителей усиливает риск появления у их потомства более выраженной патологии и свидетельствует об определенной степени неполноценности генома. Например, если у родителей имеются такие орофациальные микропризнаки (стигмы лица), как короткое небо, асимметрия крыльев носа, девиация носа, прогнатия, атипичная форма зубов, диастема, прогения и другие, то у их детей высок риск появления расщелины твердого и/или мягкого неба.
Изучение аномальных фенотипических признаков позволяет:
- составить впечатление о «мутационном грузе», лежащем на той или иной родословной;
- судить о вновь возникающих мутациях, т. е. о существовании в условиях действия мутагенных факторов;
- по ряду микроаномалий прогнозировать возможность возникновения врожденных пороков;
- объяснить накопление в относительно молодом возрасте большого числа хронически текущих заболеваний.
Отсюда следует вывод о необходимости выявления семейного наследственного предрасположения к отдельным аномалиям. Для этого важно изучить микропризнаки пороков у членов семьи пораженного (в настоящее время лучше других изучены микроформы врожденных расщелин губы и неба).
Генокопирование обусловливает чрезвычайную генетическую гетерогенность наследственных дефектов и в значительной мере осложняет генетический анализ патологических признаков.
Представленный в приложении перечень основных стигм дизэмб- риогенеза может быть использован при составлении родословных, для медико-генетического анализа, при изучении хромосомных синдромов и других наследственно обусловленных форм патологии (олигофрении, сенсорных расстройств, нервно-психических нарушений).ГЛАВА 4.
Специальные вопросы медицинской генетики 4.1. Генетика олигофрении
Выделены две клинические формы умственной отсталости: олигофрения и деменция. Термином олигофрения обозначают различные по этиологии и патогенезу состояния, объединенные двумя общими чертами: ранним возникновением интеллектуального дефекта (до трех лет) и отсутствием его прогредиентности. Олигофрения относится к числу распространенных патологий, ее частота составляет 1-3% в популяции. Деменция характеризуется распадом уже сформировавшегося интеллекта, развивается в возрасте старше трех лет. В детском возрасте такое деление достаточно условно, т. к. многие наследственные формы умственной отсталости характеризуются неравномерностью поражения и склонностью к прогрессированию.
Умственная отсталость 5
... Медико-педагогическая коррекция и профилактика умственной отсталости. 6. Вопросы экспертизы при умственной отсталости. 7. Демонстрация больных. Вводная часть. Умственная отсталость или ретардация (старый термин олигофрения) - это состояние задержанного или ...
Этиология умственной отсталости чрезвычайно разнообразна, однако в подавляющем большинстве случаев это состояние вызывается факторами, действующими внутриутробно, как наследственными, так и экзогенными. Долгое время установление этиологии умственной недостаточности отставало от других медицинских и биологических отраслей знания. Причинами умственного дефекта прежде всего стали считать инфекции и недостаточность питания. Роль наследственности в разное время оценивали по-разному. В начале XX века почти все формы умственного дефекта считали наследственными. В 30-40-е годы, наоборот, основное внимание уделяли изучению экзогенных факторов. В последнее время наряду с продолжением изучения влияния на формирование психической сферы ряда средовых, значительное место занимают исследования роли генетических факторов. Разработка методов кариологического и биохимического анализа позволила получить много ценных данных о генетических закономерностях некоторых форм умственной отсталости, понять этиологию ряда заболеваний, сочетающихся с рано развившейся умственной отсталостью.
Доля наследственно обусловленных форм олигофрении, по данным разных авторов, колеблется от 20 до 90%. Это большое различие объясняется как разными подходами авторов к оценке этиоло-
Глава 4. Специальные вопросы медицинской генетики
гической роли того или иного фактора, так и различной структурой обследуемых контингентов в отношении степени тяжести поражения. Есть основан ия считать, что наследственные факторы играют большую роль в этиологии легких степеней умственной отсталости, в то время как при тяжелых преобладают экзогенно обусловленные формы.
Актуальную задачу представляла всегда и представляет сейчас необходимая для успешной профилактики и лечения дифференциация олигофрении на отдельные клинические формы. В настоящее время, когда известны десятки средовых и генотипических факторов, вызывающих различные формы поражения интеллекта, даже самое тщательное клиническое обследование только в 1/3 случаев умственного дефекта позволяет установить с достаточной определенностью его причину. В остальной группе больных олигофренией этиология заболевания неясна или может быть указана лишь в предположительной форме.
Рассматривая причины слабоумия, можно выделить 4 группы заболеваний, одним из симптомов которых является дефект психики:
- резидуальные состояния поражения нервной системы во внутриутробном периоде, во время рождения и в первые годы постна- тальной жизни;
- первичные заболевания нервной системы;
- генные болезни;
- хромосомные болезни.
Олигофрения со специфическим синдромом поражения
Среди больных с определенным синдромом поражения значительную долю занимают экзогенно обусловленные случаи. Наследственные формы занимают меньшую долю, и соответственно этиологии представлены двумя основными группами:
Вопрос 3 Синдром умственно эмоционального перенапряжения
... стрессами сами по себе не являются вредными (патогенными). Для человека умственного труда имеет значение не столько объективная трудность производственных и учебных процессов, ... защитными приспособлениями. Способность человека к осторожности, осмотрительности проявляется в следующих формах: рациональное управление своим вниманием; правильное использование, когда необходимо, сознательного контроля ...
- больные с хромосомной патологией;
- больные с определенным моногенно наследуемым синдромом.
Хромосомная патология. К настоящему времени описано более
100 хромосомных синдромов, связанных с умственной недостаточностью. Исследования в области клинической цитогенетики показали, что геномные мутации аутосом всегда сопровождаются тяжелыми степенями умственной отсталости в сочетании с множественными врожденными пороками развития органов и систем. СамойЛ. А. Попова, Т. П. Степанова. Основы генетики в коррекционной педагогике
распространенной среди выживших детей численной аберрацией хромосом является трисомия по 21-й хромосоме, известная в клинике как болезнь Дауна. Это заболевание встречается среди новорожденных с частотой 1:700, составляя до 10% от всего контингента больных олигофренией.
Поскольку носители численных аберраций других, более крупных хромосом редко доживают даже до года, эти аберрации не имеют практически большого значения в этиологии умственной отсталости.
Структурные аномалии хромосом, возникающие в результате разрывов и последующих соединений хроматид, как правило, сопровождаются различными степенями умственной отсталости — от умеренной до глубокой — и сочетаются с множественными врожденными аномалиями, создающими иногда довольно специфический образ «олигофрена-диспластика». Наиболее существенными признаками при этом являются: сочетание умственной отсталости с отставанием в физическом развитии и множественными соматическими аномалиями (краниофациальная дисплазия, ненормальная форма и расположение ушных раковин, гипертелоризм и эпикант, готическое небо, аномалии строения глазных щелей и яблок, специфическое изменение кожного рисунка на ладонях и подошвах, аномалии строения и расположения пальцев рук и ног и др.).
В литературе в последние годы появились описания многих наследственных синдромов, одним из симптомов которых является олигофрения. Характер ци- тогенетических аномалий при большинстве из этих синдромов требует дальнейших уточнений.
Синдром Корнели и де Ланге. Выраженная врожденная гипотрофия, значительная задержка физического и умственного развития, грубые пороки развития. Описаны частичные трисомии, а также трансплокации, инверсии различных хромосом. Большинство случаев синдрома — спорадические.
Синдром Прадера — Вилли характеризуется мышечной гипотонией, половым недоразвитием, ожирением и умственной отсталостью. Кариотипически — делеция 15-й хромосомы. Дефектная хромосома всегда имеет отцовское происхождение. Синдром Ангельмана характеризуется дефектом 15-й хромосомы материнского происхождения.
На основании существующих данных нельзя с точностью судить о том, какую долю среди олигофренов составляют лица с различными структурными и числовыми аутосомными аберрациями. Однако
есть основания предполагать, что, кроме болезни Дауна, аутосомные аберрации не имеют существенного значения в этиологии олигофрении вследствие редкости синдромов и значительной ограниченности продолжительности жизни страдающих ими детей.
Олигофрения
... хромосом; моногенные, возникающие в связи с изменениями в одном гене; синдромы умственной отсталости с неуточненным типом наследования. Допускается, что среди последних ... опорно-двигательного аппарата, речи); олигофрения с психопатоподобными формами поведения; олигофрения с выраженной лобной недостаточностью. Среди клинических классификаций олигофрении большое место занимают этиологические, ...
Общее нарушение генного баланса, связанное с избытком или недостатком дозы генов, расположенных в половых хромосомах, гораздо менее фатально отражается на развитии организма, чем это имеет место при аутосомных аберрациях. Поэтому наличие гоносом- ных аберраций совместимо не только с рождением, но и с нормальной жизнеспособностью и даже, в ряде случаев, с нормальным фенотипом. Умственная отсталость далеко не всегда сопровождает го- носомные мутации.
Согласно статистическим данным, 17-25% мужчин с синдромом Клайнфельтера имеют сниженный интеллект. Лишняя Х-хромосо- ма у женщин проявляется в еще большем снижении интеллекта, чем у мужчин. Отмечается прямая корреляция между числом лишних Х-хромосом и степенью интеллектуального снижения. Синдром Шерешевского — Тернера более редок среди умственно отсталых женщин, составляя в среднем, по данным разных авторов, 0,6:1000.
Причины умственной отсталости при аутосомных и гоносомных аберрациях, очевидно, лежат в грубых нарушениях генного баланса, как следствие нарушения множества ферментных функций, приводящих к искажению эмбрионального развития. Это обстоятельство обусловливает малую эффективность лечения олигофрений, связанных с хромосомными аберрациями, и заставляет уделять основное внимание изучению причин их возникновения и возможной профилактике. Согласно статистическим данным, больные с хромосомными аномалиями составляют около 15% всех случаев слабоумия.
Моногенно наследуемые олигофрении со специфическим синдромом поражения
В группе олигофрений с определенной клинической картиной ведущее место занимают ферментопатические формы, включающие нарушения всех видов обмена веществ: белкового, углеводного, ли- пидного, минерального. Учение о наследственных нарушениях обмена веществ берет свое начало от исследования английского ученого Кэррода, который в 1906 году при изучении четырех форм на-JI. А. Попова, Т. П. Степанова. Основы генетики в коррекциониой педагогике
рушения обмена веществ у детей (алкаптонурии, альбинизма, пен- тозурии и цистинурии) высказал предположение о том, что в основе указанных страданий лежит дефект фермента наследственной природы. Эта мысль получила конкретное биохимическое обоснование и всеобщее признание лишь несколько десятилетий спустя, и такого рода заболевания получили название ферментопатий (энзимопатий).
К настоящему времени известно несколько сотен наследственных нарушений обмена веществ. При большинстве из них происходит поражение нервной системы, что приводит к возникновению так называемого сложного дефекта — сочетанию интеллектуальной недостаточности с нарушениями зрения, слуха, опорно-двигательного аппарата.
Другая группа моногенно наследуемой умственной отсталости диагностируется по наличию своеобразного соматического или неврологического синдрома. Описано большое количество олигофрений с глазными изменениями: микроктальмией, катарактой, аниридией; с кожными патологиями: меланолейкодермией, ихтиозом, телеанги- эктазиями, с различными дизостотическими нарушениями и т. п.
Многие из моногенно наследуемых форм олигофрении представляют редкие своеобразные синдромы. Среди моногенно наследуемых олигофрений самыми частыми, но мнению исследователей, являются нейрофиброматоз Реклингаузена и миопатия Дюшенна. Исследования последних десятилетий показали, что умственная отсталость, сопровождающая эти заболевания, является непрогре- диентной, несмотря на прогрессирование основной симптоматики болезни, что позволяет отнести интеллектуальный дефект при этих заболеваниях к олигофрении.
Свыше шестидесяти генов Х-хромосомы могут определять умственную отсталость, чаще — в сочетании с нарушениями других органов и систем организма (синдромы Арского, Коффина — Лоури, Борьесона — Форсмана — Аемана, Дюшенна, Гольдблатта, Гольтца, Лоу, Норри, Ленца, Ретта, Леша — Нихана).
Наследоваться они могут по рецессивному (чаще) и доминантному типам.
Синдром Нунан. Аутосомно-доминантный тип наследования. Частота синдрома среди новорожденных 1:1000-1:2500. Впервые описан в 1928 году Вейсенбергом. К1963 году описана большая группа больных низкого роста, с нарушениями в развитии скелета, птери- гиумом, страдавших врожденными пороками сердца. По внешнему
виду эти люди походили на больных с синдромом Шерешевского- Тернера, однако среди них были и лица мужского пола. Поскольку кариотип этих больных соответствует норме, синдром Нунан получил название «тернеровский фенотип с нормальным кариотипом». Дети отстают в росте, психомоторном и особенно речевом развитии, характерна задержка полового развития. Особенности поведения напоминают психопатические проявления при раннем детском аутизме. Умственная отсталость наблюдается в 60% случаев, нередки дефекты зрения: миопия, кератоконус, косоглазие.
Синдром Рубинштейна — Тейби. Предположительно аутосомно- доминантный тип наследования с неполной пенетрантностью.
Синдром Дубовица. Впервые описан в 1965 году. Характеризуется врожденной гипотрофией, отставанием физического развития в по- стнатальном онтогенезе. Аутосомно-рецессивный тип наследования.
Синдром Барде — Бидля. Одно из нервно-психических заболеваний, сочетающихся с ожирением. Наблюдается прогрессирова- ние интеллектуального дефекта. Тип наследования аутосомно-ре- цессивный.
Синдром Гольтца. Наследуется по Х-сцепленному доминантному типу с летальностью для плодов мужского пола. Все девочки умственно отсталые с нарушениями зрения, а в некоторых случаях и слуха.
Синдром Опица — Каведжиа. Впервые описан в 1974 году. Тип наследования Х-сцепленный рецессивный. У всех больных мальчиков диагностируется умственная отсталость, нередки нарушения зрения (косоглазие, птоз) и слуха (нейросенсорная тугоухость).
Синдром Беквита — Видемана. Описан в 1963 году. Частота среди новорожденных 1:1200. Тип наследования аутосомно-доминан- тный. Умственная отсталость отмечается у 12% больных. Уже при рождении отмечается гипогликемия, сочетающаяся с рядом соматических изменений. Гигантизм отмечается либо с рождения, либо развивается постнатально. Возможна микро- и гидроцефалия, мак- роглоссия, врожденные пороки сердца, диафрагмальные грыжи, незавершенный поворот кишечника, висцеромегалия (увеличение печени, селезенки и др.).
Две рассмотренные группы олигофрений, обусловленные хромосомными аберрациями и моногенно наследуемые со специфическим синдромом поражения, составляют небольшую долю среди всехЛ. А. Попова, Т. П. Степанова. Основы генетики в коррекционной педагогике
случаев олигофрении. Более важную и сложную проблему представляет генетика клинически недифференцированных форм, которые составляют большинство среди больных олигофренией.
Клинически недифференцированные формы олигофрении
Недифференцированная олигофрения — олигофрения, не сопровождающаяся явными соматическими или неврологическими изменениями, либо определенными ферментными дефектами, или хромосомными аберрациями. Такие формы представляют наиболее острую проблему генетики олигофрении, что связано с их широкой распространенностью и малой изученностью этиологических факторов при этих формах. Трудность изучения этой группы олигофре- ний усугубляется тем, что одну и ту же клиническую картину могут вызывать различные факторы, как наследственные, так и экзогенные. Все зарубежные исследователи основную этиологическую роль в возникновении недифференцированной олигофрении отводят эндогенным, т. е. генетическим, факторам. Тому есть целый ряд доказательств.
- Установлена высокая (80-100%) конкордантность по олигофрении однояйцевых близнецов.
- Отмечается повышенная частота кровнородственных браков среди родителей больных.
- Описаны факты отчетливого семейного накопления олигофрении.
Исследователи выделяют две группы клинически недифференцированной олигофрении. Первую группу составляют более тяжелые случаи олигофрении у детей интеллектуально полноценных родителей, вторую, более многочисленную — случаи олигофрении с меньшей степенью дефекта и со значительным семейным отягощением. Роль генетических факторов в этиологии олигофрении у больных первой группы сводится, по-видимому, к действию редких рецессивных аллелей. Среди родителей этой группы больных наблюдается повышенная частота кровного родства. Эти тяжелые моногенно наследуемые формы клинически недифференцированной олигофрении относительно редки.
Семейная недифференцированная олигофрения. Различные исследования показали, что 30-50% пробандов с недифференцированной
олигофренией имеют одного или обоих умственно отсталых родителей. Некоторые авторы недифференцированную олигофрению у детей в таких семьях связывают с плохой семейной обстановкой. Известно, что именно в этой группе больных накапливаются и биологические экзогенные вредности, которые сами по себе могут быть причиной олигофрении: частые заболевания детского возраста и другие вредности внутриутробного периода,в частности, алкоголизм матерей. Нельзя исключить возможность средовой передачи олигофрении от одного поколения к другому. В семьях подобного рода отмечается увеличение числа детей с задержкой психического развития. Однако большинство исследователей считает, что доля генетических факторов в возникновении семейной недифференцированной олигофрении превышает долю отрицательных влияний среды, хотя каждый фенотипический признак, а тем более такой сложный, как интеллект, является продуктом взаимодействия наследственности и среды, в которой формируется.
Из генетических гипотез при семейной недифференцированной олигофрении наибольшую популярность имеет гипотеза полигенов аддитивного (от лат. additivus — прибавляемый) действия. При большом числе аддитивно действующих генов распределение фенотипов будет соответствовать нормальному, где на одном конце будут лица с наиболее высоким интеллектуальным уровнем, а на другом — с наиболее низким. Сопоставление ожидаемого (на основании полигенной гипотезы) распределения популяции по интеллектуальному уровню с фактическим указывает на соответствие для легких форм олигофрении и значительные отличия для тяжелых.
Для объяснения характера наследования недифференцированной олигофрении существуют и другие генетические гипотезы. Так, например, предполагается, что 1 /3 случаев недифференцированной олигофрении средней тяжести вызвана рецессивными генами (их насчитывают несколько десятков), локализованными в разных хромосомах. Около трети лиц всей популяции являются гетерозиготными носителями таких генов. В гомозиготном состоянии эти рецессивные гены снижают интеллект в различной степени.
Хотя генетические гипотезы происхождения олигофрении имеют целый ряд серьезных подтверждений, все же они остаются гипотезами. Вопрос о том, чему принадлежит ведущая роль в этиологии семейной недифференцированной олигофрении — наследственности
1